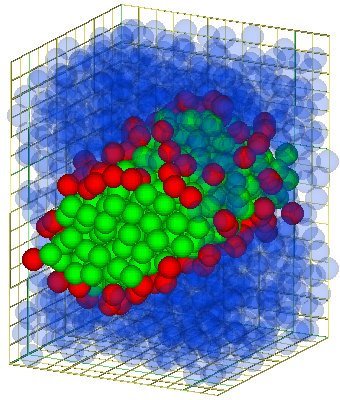
 |
Computer Simulations of Surfactant-Water-Systems |
|
Surfactant-water systems may be described in different ways, either
- in an atomic description, as is it done several simulation, or
- by continuum theories, such as elasticity theory.
The validity of the first comes from the knowledge of the atomic interactions while the elasticity theory (e.g. the Helfrich Hamiltonian) has been proof right by a huge amount of experiments. However, The atomic description is valid up to scales of some nanometers. On the other hand the experiment proof right the elasticity theory down to some 100 nanometers. Hence there is an open gap in length scales in the description of such systems.
The model
The goal of my work is now to close this gap. Since atomically detailed simulation, like those of Berendsen [1], are to CPU-time consuming to reach the necessary system sizes and simulated times, I used a simplified, effective model. This model is similar to a model introduced by B. Smit [2] in the early 90's. Only four types of particles are distinguished:
- water-particles (hydrophilic), denoted by w,
- oil-particles (hydrophobic), denoted by o,
- headgroup-particles (hydrophilic), denoted by h or by H and
- tail-particles (hydrophobic), denoted by t or by T.
This particles interact via repulsive softcore-potentials if one of the particles is hydrophilic and the other hydrophobic. In all other cases the interaction-potential is a attractive Lennard-Jones-potential. To create molecules containing more than one particles we add a harmonic bond potential and in the most cases a bending rigidity along the chain. In the latter case the capital letter are used to denoted the particles. The following picture shows the most commonly used surfactants:
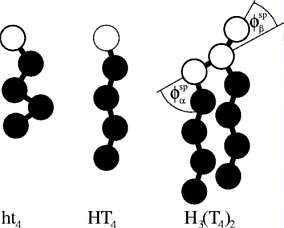
The simulations
Using this model a couple of MD-simulation where carried out to investigate
- the selfassembly of the model surfactants and
- the properties of bilayers made out of these surfactants. Of special interest were the following properties:
- lateral and transversal diffusion,
- the stretching elasticity and
- the bending elasticity.
Conclusion
It has been shown that that the model surfactants selfassembly into micelles and bilayers, depending on the concentration. Furthermore this bilayers are in fluid state. The investigation of this bilayers shows that the elasticity theory is valid down to a lateral length of several surfactant molecules. This is in physical units of the order of 2 nanometers.
This work was done together with my supervisor Prof. Lipowsky. I have to thank Dr. Gompper for many helpful discussions.
Literature:
- E.Egberts, H.J.C Berendsen; J. Chem. Phys. 89 (1988); 3718-3732
- B.Smit, P.A.J. Hilbers, K.Esselink, L.A>M. Rupert, N.M. Van Os, A.G. Schlijper; J.CHem. Phys. 95 (1991); 6361-6368
- Read more about this topic in the report about the Research Activities 1995/96 of the theory division of the MPI Golm (used to be in Teltow).
- R. Goetz; PhD-thesis, Universität Potsdam 1997.
- R. Goetz, R. Lipowsky; J. Chem. Phys. 108, (1998), 7397-7409
- R. Goetz, R. Lipowsky; Phys. Rev. Letters 82, (1999), 221-224